An approach to evaluate therapeutics program quality
10 mins to read
January 16, 2024
Picking the right therapeutics programs at the right companies is critical to investors, and understanding investor expectations is essential to entrepreneurs who want to see their products mature to benefit patients. Plenty of publications describe the cross-industry predictors of startup success, like quality of the founding team and operational rigor. But throughout my career as an academic, entrepreneur, and investor, I’ve found a dearth of materials answering the field-specific question: “What makes a good therapeutics development program?”
This article outlines my hypothesis-driven approach for evaluating the quality of any given therapeutics program as one component of the larger drivers of success for a company. Using this framework, a program is viable for development if all hypotheses outlined in Figure 1 are true. This methodology is not the only solution, but I hope its availability will help academics looking to translate promising discoveries, early-career entrepreneurs who want to ensure their assets will stand up to scrutiny, and new venture associates developing their diligence process.
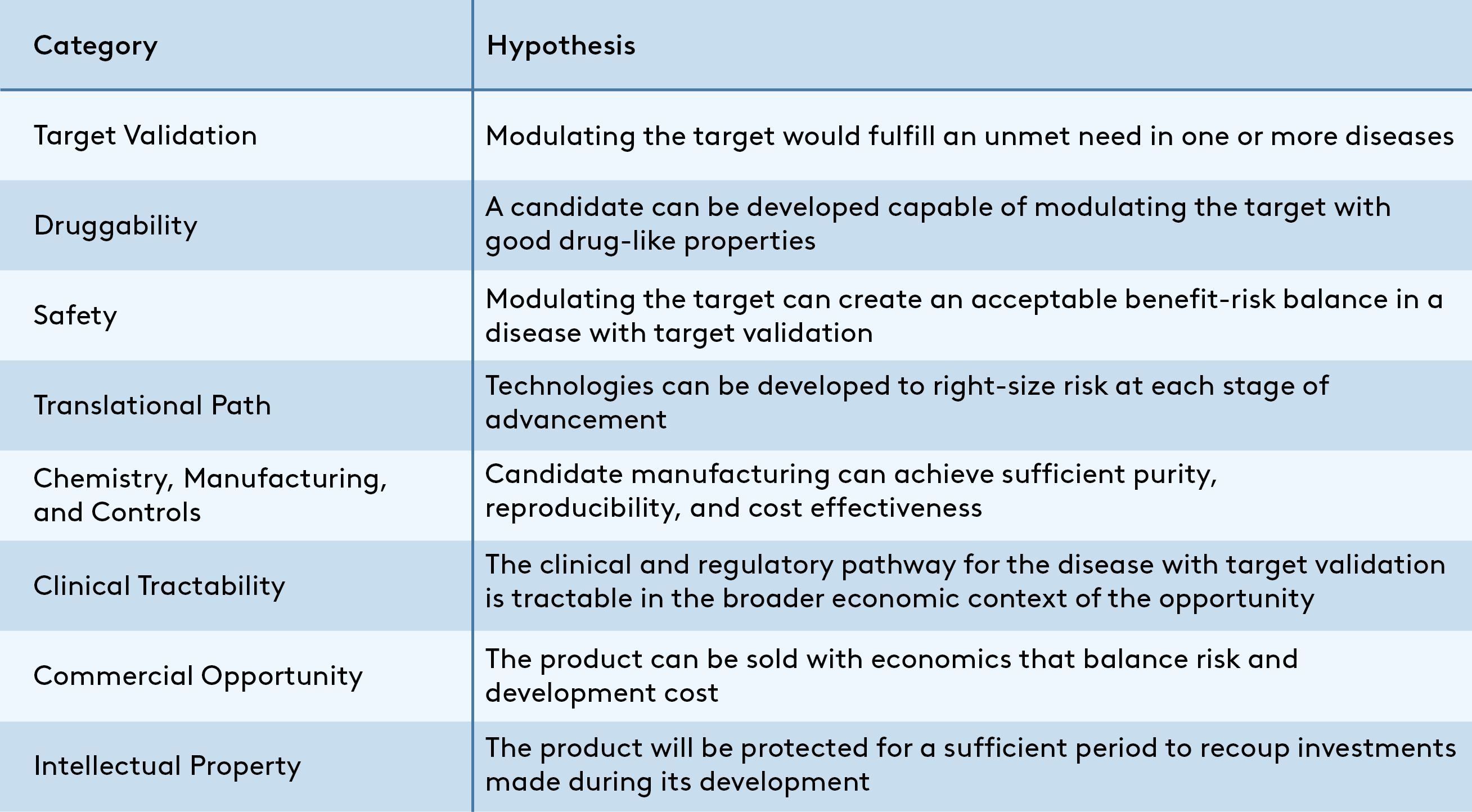
A program is viable if all hypotheses are true, but an integrated assessment is essential to differentiate “viable” from “potentially transformative” programs. A spark that makes a program stand out could come from revolutionary biology rooted in deep insight into unmet need, herculean academic efforts to create a drug-like molecule in a chemically challenging but sought-after class, or even from outside this asset-focused framework in the form of a proven, energetic team. On the other side of the coin, it is rare to find a program without “warts” in the form of one or more uncertain hypotheses that should be de-risked as the program advances. In each case, experience-based judgment is critical to convert evaluation of a program’s strengths and weaknesses into a useful business decision.
While it’s impossible to detail all considerations that might support or refute the hypotheses from Figure 1 in a single post, some examples of the types of data and analyses that can support each are provided as a primer below.
Target Validation
Conviction that modulating the target will address an unmet need is table-stakes for a therapeutics program. A compelling data package will often center on experiments in an animal disease model. This may be genetic interventions for a target-to-hit program, or pharmacologic modulation with a lead compound at later stages. In either case, the ideal experiment will mirror the way a patient might use a therapeutic arising from the program. For example, if patients are typically not aware of their disease until it reaches an advanced stage, the most compelling experiments will treat animals with a fully established disease. In the strongest data package, the in vivo foundation is supported by a variety of orthogonal experiments pointing toward the same conclusion. In vitro experiments using patient-derived samples and real-world data such as GWAS may, for example, suggest the observations in mouse models would translate to the human condition.
Druggability
The ability to modulate a target in a therapeutically useful way is among the greatest challenges in biopharma. One needs not only inhibit a cellular activity, but access the relevant tissues, for an appropriate length of time between dosing, and without affecting other critical biological processes. For Hit-stage and later programs, characterization of the most advanced asset (e.g., potency, ADME, PK, etc.) will identify the specific challenges that must be overcome to achieve a drug-like candidate. For pre-Hit programs, historical precedent for similar targets will indicate feasibility. For example, small molecule drugs have been FDA approved to inhibit many enzymes with an accessible catalytic pocket, but targeting transcription factors or scaffolding proteins may require newer, more challenging modalities that elevate cost and risk.
Safety
Safety remains a risk to any drug until years of post-market data is available, and the acceptable profile will vary based on severity of the disease targeted and the effect size of the drug. However, guideposts exist that may indicate the magnitude of that risk in the context of a given development plan. For early-stage programs, knockout mice and GWAS associations may differentiate a target with minimal overt on-target consequences suitable as a preventive agent from a target where liabilities will likely limit use to severely ill populations. For Lead and Candidate stage programs, careful design of chronic in vivo proof-of-concept experiments can enhance confidence in tolerability for the specific asset; if there is a known liability risk, for example, the relevant tissue may be harvested and examined for abnormalities alongside efficacy readouts. For later stage programs, standard GLP-Toxicology and clinical experience provide more robust confidence in acceptable tolerability for an asset.
Translational Path
One of the most frequent, expensive failure points in drug development is the Phase 2 trial, where only ~30-50% of drug candidates manage to show an efficacy signal in patients. Many factors contribute to Phase 2 failures, but among the most common explanations are:
• Poorly predictive animal models: For monogenic diseases excellent mouse models are often available; targeted mutation in the causative gene may recapitulate the disease phenotype, and clinically proven treatments may even confirm their predictive validity. However, preclinical models are of questionable translational value for certain complex diseases. In these cases, heightened risk can sometimes be partially mitigated by innovative technologies such as patient derived organoids, but development cost and uncertainty will remain greater.
• Sub-optimal dosing: Converting an efficacious animal dose to an equivalent human dose is not straightforward, and even sophisticated pharmacokinetic models incorporating differences in bioavailability, protein binding, and stability are imperfect. A target engagement biomarker, which allows direct confirmation that a drug is exerting its desired biological effect, is a critical tool to minimize expensive phenotypic dose-finding studies.
While de novo technology development can sometimes address translational challenges, the landscape of pre-existing technology sets baseline risk and required investment arising from this category.
Chemistry, Manufacturing, and Controls (CMC)
At-scale manufacturing of a never-before-synthesized drug product is not trivial! The product must be pure, reproducible, stable during storage, and sufficiently inexpensive that the unit economics will be favorable once commercialized. Required CMC expertise and investment should not be underestimated for a small molecule, but biologics and newer modalities such as gene therapy are a special challenge. Traditional small molecules and antibodies will typically not require process optimization before candidate selection, but more nascent modalities such as cell therapy will benefit from in-house expertise early in the development process. Special attention may be paid to manufacturing costs in the context of modality and target indication. For example, an autologous cell therapy which costs tens of thousands of dollars per dose to manufacture would be inconsistent with a preventive indication where benchmark pricing is <$20K annually, but could be acceptable in oncology where benchmarks range upwards of $100K.
Clinical Path
The challenge of demonstrating clinical efficacy will differ by disease across several axes, including:
• Regulatory uncertainty, which is driven by the existence of precedent. For example, multiple drugs have been approved for rheumatoid arthritis and the FDA has published guidance documents that clearly outline expectations for future therapies. For other indications, no therapeutic has yet reached Phase 3 clinical trials, and acceptable endpoints will need to be defined in consultation with regulators.
• Feasibility, which is largely determined by availability of patient populations to the drug developer; patients may be concentrated in a few centers or spread across many, or multiple clinical-stage companies may be competing to engage a limited pool of patients for their trials.
• Resource intensity, which is most influenced by the current standard of care, the magnitude of effect expected for the therapeutic, and the rate at which the study endpoint manifests. For example, standard of care for some diseases like diffuse large B cell lymphoma achieve cure rates of >75%, meaning large trials are required for even a 90% effective therapeutic to demonstrate a statistically significant difference. Similarly, the impact of a drug on some diseases can be assessed in weeks via a test of functional capability; in others, participants must be followed for years to observe the clinical outcomes of treatment.
Commercial Opportunity
It is an unfortunate reality that low commercial potential impedes for-profit investment in many diseases with high unmet need. This phenomenon is best illustrated by the category of “neglected tropical diseases”, which primarily affect impoverished populations and remain largely untreated due to inability to recoup investment from patients. One worst-case scenario for a drug developer is to incur substantial expense to advance through Phase 1 trials yet fail to reach patients because poor commercial opportunity compromises their Phase 2 and Phase 3 investment case. Key drivers of commercial opportunity include pricing, size of the indicated population, and the commercialization force necessary to drive sales. These factors, which are among the largest drivers of net profitability, should be weighed against development costs to ensure that economic returns are possible. A risk-adjusted net present value analysis can integrate the dozens of assumptions necessary for this comparison, and can directionally indicate the commercial opportunity of a program or highlight key risk areas.
Intellectual Property
For-profit pharmaceutical investors require substantial future cashflows to compensate for the high cost and risk of development. Even with good unit economics, generating sufficient cashflows is only possible by obtaining exclusivity – a time period during which the developer holds a monopoly on sales of the drug. The most robust exclusivity mechanism is a composition of matter patent, which protects the structure of the drug and forms the core of most intellectual property portfolios. While a granted patent is not essential for most pre-Candidate programs, a clear path to obtaining one is. As the program advances a portfolio of additional patents will ideally provide supplementary protection, often covering formulations, dosing, and other methods of use. Regulatory exclusivity mechanisms such as the seven years attached to orphan drug approval can supplement a patent portfolio, but their length is rarely sufficient to protect an investment on their own.
A more detailed overview of intellectual property considerations created by true experts is available free-of-charge through one of the world-leading legal firms, Morrison Foerster.
Enacting this approach can drain substantial organizational resources if analyses are not carefully managed. In my experience, a balance of efficiency and rigor can be achieved by progressing the effort through stages of increasing analytical depth targeting the most uncertain hypotheses first. For example, one might complete a “pre-assessment” to highlight potential areas of weakness in the program. Then in a second stage, one might engage technical experts in the highlighted areas to answer tailored questions that can confirm or refute those weaknesses. Ultimately, the appropriate depth to stop analysis will be determined by its purpose; a clinical-stage acquisition requires a lot more certainty than a discovery-stage portfolio prioritization decision! And while this article is not intended to enable the reader to conduct a rigorous diligence all on their own, I hope it will provide a primer to the essential components.
If you found this post helpful, please reach out and let me know which topics you’d like to learn more about! And if you have recommendations on how to adjust or improve this framework, let me know that as well!
About the author
Ethan Sarnoski, PhD is a Senior Ventures Director at Cambrian Biopharma, where he works with field-leading scientists to translate discoveries in the biology of aging into actionable drug development programs for aging-related disease. He has 10+ years of experience as a molecular biologist, management consultant, entrepreneur, and investor. You can contact him via LinkedIn at https://www.linkedin.com/in/ethansarnoski/.